109. Interpretazione dei fenomeni elettrolitici. — Dopo una serie d’investigazioni importantissime noi possediamo oggi una teoria più che soddisfacente dei fenomeni elettrolitici, dovuta specialmente a Clausius, Helmholtz, Arrhenius e Nernst. Esporremo un breve cenno delle idee adesso dominanti, tacendo per brevità sul lento sviluppo storico che le ha condotto alla perfezione attuale.
Noi abbiamo detto, parlando delle soluzioni (vol. 1° § 94), che nel seno del solvente vagano liberamente le molecole del corpo sciolto, così come le molecole d’un gas vagano liberamente nel vaso che lo contiene. E si corrispondono perfettamente, nei rapporti concettuali e sperimentali, la pressione osmotica delle soluzioni, misurabile col metodo del setto semipermeabile, e la pressione del gas.
Riferimmo pure che soluzioni contenenti egual numero di molecole per litro possiedono la stessa pressione osmotica, così come gas differenti, alla stessa temperatura, e contenenti egual numero di molecole per litro, esercitano la stessa pressione sulle pareti (leggi di Van t’Hoff e d’Avogadro).
Questa legge non è però rigorosa nel caso delle soluzioni; e mentre si verifica bene per alcune soluzioni, come quella di zucchero, non avviene lo stesso per le soluzioni acquose degli acidi, delle basi o dei sali, cioè proprio per quelle soluzioni che sono conduttrici dell’elettricità e si decompongono per il suo passaggio. La legge di Van t’Hoff è adunque in difetto negli elettroliti.
Questo singolare comportamento degli elettroliti suggerì ad Arrhenius e a Nernst l’ipotesi dalla dissociazione elettrolitica, che rende conto, insieme, delle deviazioni dalla legge di Van t’Hoff e della conducibilità degli elettroliti. Si ammette in questa ipotesi che nella soluzione di un elettrolito parte delle molecole vaganti nel liquido sono spezzate, o dissociate, in due semi-molecole cariche l’una d’elettricità positiva e l’altra d’elettricità negativa. Così in una soluzione di K Cl esisterebbero delle molecole neutre, e insieme degli ioni positivi K+ e degli ioni negativi Cl-provenienti dalla dissociazione di altrettante molecole K Cl. Questi ioni si muovono in tutti i sensi nel seno del liquido; talvolta si ricombinano, mentre altre molecole si scindono, ma in modo tale che per una data soluzione, a una data temperatura, una frazione costante delle molecole totali del corpo sciolto sarebbero dissociate.
Gli ioni vaganti, come il K+ e il Cl- non son da confondere con gli atomi corrispondenti di K e di Cl, poichè sono atomi provveduti di cariche elettriche energiche; ed è appunto in virtù di queste cariche che possono esistere nell’acqua gli ioni di potassio, mentre l’atomo di potassio, come sappiamo, reagisce vivamente col liquido. Solo quando, con un mezzo qualsiasi, gli ioni perdono le loro cariche elettriche, si trasformano in atomi chimici, e riprendono le proprietà chimiche di questi.
Si riconosce subito, intanto, che il numero totale di individualità vaganti nel liquido viene con ciò accresciuto; poichè se nell’unità di volume del solvente sono state introdotte, ad esempio, 1 milione di molecole del sale, e la metà di queste si son dissociate, esisteranno nel liquido 500.000 molecole neutre, 500.000 ioni K+ e 500.000 ioni Cl-, in totale perciò vagheranno nel liquido 1 milione e mezzo di particelle o gruppi indipendenti, determinando così una pressione osmotica una volta e mezza maggiore di quella spettante alla totalità delle molecole, qualora non fossero dissociate. E s’intende, inoltre, che dall’entità delle deviazioni rispetto alla legge di Vant’Hoff si potrà dedurre la frazione di molecole dissociate, che esercitano perciò una pressione parziale doppia della normale.
La presenza di questi ioni che portano, qualunque sia la sostanza sciolta, una carica elettrica determinata, se la molecola si scinde liberando una valenza (K—Cl = K+ + Cl-), o una carica doppia della precedente se si scinde liberando una doppia valenza:
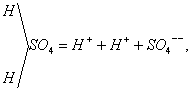
spiega ancora la conducibilità elettrica degli elettroliti e le leggi dell’elettrolisi.
Quando, infatti, s’introducono in una soluzione due lamine metalliche (gli elettrodi) rilegati ai poli d’una pila (fig. 138), si crea tra le due lamine un campo elettrico sensibilmente uniforme, come tra le due armature d’un condensatore piano.
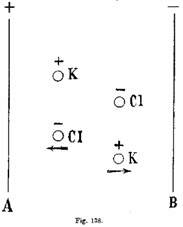 Sotto
l’azione di questo campo il movimento degli ioni carichi, che si compiva prima
indifferentemente in tutte le direzioni, viene perturbato; l’ione K+
è sottoposto invero a una forza nel senso AB, e l’ione Cl- a una
forza eguale ed opposta nel senso BA. Il moto conseguente sarebbe uniformemente
accelerato se l’ione non si trovasse in un mezzo che, per attrito, si oppone al
movimento; invece, in virtù dell’attrito, presto si raggiungerà l’equilibrio
dinamico, e gli ioni
assumeranno, come le gocce di pioggia nell’aria, un moto uniforme; questo sarà
tanto più veloce quanto maggiore è la intensità del campo, e maggiore la mobilità
dell’ione, cioè minore l’attrito col mezzo. Appena gli ioni positivi
giungono sulla lamina B, e i negativi sulla A, cedono alle lamine le loro
cariche elettriche, trasformandosi in atomi chimici con tutte le loro proprietà; ma in conseguenza di questa
precipitazione convettiva di cariche elettriche opposte sulle lamine A e B, la
differenza di potenziale tra queste tende a diminuire.
Sotto
l’azione di questo campo il movimento degli ioni carichi, che si compiva prima
indifferentemente in tutte le direzioni, viene perturbato; l’ione K+
è sottoposto invero a una forza nel senso AB, e l’ione Cl- a una
forza eguale ed opposta nel senso BA. Il moto conseguente sarebbe uniformemente
accelerato se l’ione non si trovasse in un mezzo che, per attrito, si oppone al
movimento; invece, in virtù dell’attrito, presto si raggiungerà l’equilibrio
dinamico, e gli ioni
assumeranno, come le gocce di pioggia nell’aria, un moto uniforme; questo sarà
tanto più veloce quanto maggiore è la intensità del campo, e maggiore la mobilità
dell’ione, cioè minore l’attrito col mezzo. Appena gli ioni positivi
giungono sulla lamina B, e i negativi sulla A, cedono alle lamine le loro
cariche elettriche, trasformandosi in atomi chimici con tutte le loro proprietà; ma in conseguenza di questa
precipitazione convettiva di cariche elettriche opposte sulle lamine A e B, la
differenza di potenziale tra queste tende a diminuire.
E se la pila la mantiene costante, deve restituire alla lamina A la perduta elettricità positiva, e sottrarre alla B l’acquistata elettricità positiva, cosicchè un flusso continuo d’elettricità positiva avrà luogo dal polo positivo della pila verso A, e da B verso il polo negativo della pila; cioè una corrente nel senso relativo circolerà lungo i fili che rilegano il voltametro alla pila, ed essa si potrà anche considerare come avente luogo nel voltametro, ove avrà però carattere convettivo.
Raddoppiando il campo elettrico tra A e B si raddoppierà la velocità costante di regime assunta dagli ioni; un numero doppio di questi giungerà nello stesso tempo sugli elettrodi, e si raddoppierà perciò l’intensità della corrente nel circuito che rilega la pila al voltametro. Malgrado il carattere convettivo della corrente, gli elettroliti obbediranno perciò alla legge di Ohm, come i conduttori metallici, e si potrebbe anche misurare in Ohm la loro resistenza dalla differenza di potenziale occorrente per produrre la corrente uno, se non intervenisse una speciale perturbazione agli elettrodi di cui diremo più in là. La resistenza, misurata con metodi opportuni, risulta, come nei metalli, proporzionale alla lunghezza del conduttore elettrolitico, e inversamente proporzionale alla sua sezione.
Un’influenza notevole sulla conducibilità viene esercitata
dalla concentrazione della soluzione. Se la frazione di molecole
dissociate fosse sempre la stessa, per qualunque concentrazione, allora
raddoppiando il numero di molecole sciolte si raddoppierebbe il numero di
quelle dissociate, e quindi degli ioni presenti. E poichè al numero di questi è
proporzionale la carica trasportata sugli elettrodi sottoposti a una data
differenza di potenziale, sarebbe anche raddoppiata la conducibilità della
soluzione. L’esperienza ha però rivelato che la frazione di molecole
dissociate sul loro numero totale è tanto minore quanto più la soluzione è
concentrata. Se perciò a una certa concentrazione sono dissociate la metà delle
molecole sciolte, a una concentrazione doppia ne saranno dissociate, per
esempio, solo i ![]() e perciò il numero di ioni esistenti sarà maggiore
di quello di prima, ma non sarà il doppio di esso, e perciò la conducibilità
diverrà un po’ meno del doppio. Si intende con ciò come dall’andamento della
conducibilità di una soluzione, di cui si vari la concentrazione, si possa
dedurre la frazione del numero di molecole dissociate sulle totali disciolte.
Lo stesso calcolo può farsi con le deviazioni della legge di Van-t’Hoff sulla
pressione osmotica, come pure con le deviazioni dalle leggi di Raoult
sull’alterazione dei punti di solidificazione o di ebollizione del
solvente, per virtù del corpo disciolto, deviazioni che possono essere
egualmente interpretate. Con questi diversi metodi, malgrado i fenomeni
disparati con cui si ha da fare, il valore calcolato della frazione di
dissociazione è sensibilmente lo stesso. Si è
così trovato che solo in una soluzione estremamente diluita tutte le
molecole esistenti sono dissociate, e che invece, aumentando la concentrazione,
la frazione delle dissociate sulle totali va sempre diminuendo.
e perciò il numero di ioni esistenti sarà maggiore
di quello di prima, ma non sarà il doppio di esso, e perciò la conducibilità
diverrà un po’ meno del doppio. Si intende con ciò come dall’andamento della
conducibilità di una soluzione, di cui si vari la concentrazione, si possa
dedurre la frazione del numero di molecole dissociate sulle totali disciolte.
Lo stesso calcolo può farsi con le deviazioni della legge di Van-t’Hoff sulla
pressione osmotica, come pure con le deviazioni dalle leggi di Raoult
sull’alterazione dei punti di solidificazione o di ebollizione del
solvente, per virtù del corpo disciolto, deviazioni che possono essere
egualmente interpretate. Con questi diversi metodi, malgrado i fenomeni
disparati con cui si ha da fare, il valore calcolato della frazione di
dissociazione è sensibilmente lo stesso. Si è
così trovato che solo in una soluzione estremamente diluita tutte le
molecole esistenti sono dissociate, e che invece, aumentando la concentrazione,
la frazione delle dissociate sulle totali va sempre diminuendo.